
It's time to split: a case for phenotype-wise enrollment
We must drop current sepsis definitions.

CRITICAL CARE is a peculiar field. Perhaps due to the need to be operational at the bedside, we have created disease mimics to work with. Of course, I am talking mainly about ARDS and sepsis.
In the last couple of years, Dr Lynn and I have been campaigning against what we named “The Petty and Bone Mistake”, and “The Petty and Bone RCT”, after the deceased prominent intensivists-researchers Thomas Petty and Roger Bone, early promoters of what we now call ARDS (Petty) and sepsis (Bone) definitions.
They were lumpers, that is, they believed patients could be grouped for treatment and research if they had a similar clinical presentation, whatever caused the illness. The results are the well-known bedside, cause-agnostic triage tools that were ultimately named “definitions" for ARDS and sepsis.
To be fair, they had a good guess. Petty saw an analogy with the newborn respiratory distress syndrome due to lack of alveolar surfactant, and Bone was among those early promoters of the generic and quasi-metaphysical “dysregulated inflammation” mantra, whatever it means. To understand what went wrong, we will need medical epistemology lenses. Yes, dear reader. You only find it here.
The Petty and Bone paradigm is dead

Once more, top critical care researchers will reaffirm the death of the Petty and Bone paradigm - and refuse to see it.
There are many ways one can define what a disease is, but in critical care, illnesses pertain to the realm of human biology. There must be a mechanistic biological model, or what I call a disease model, underpinning the nosologic entity. It must point to a dominant cause, and it has to be a testable conjecture, in a Popperian sense.
Petty's ARDS and Bone's sepsis are failed models. They couldn't predict events in the empirical world. Let's simplify the language. The disease models were tested in RCTs and failed tens, hundreds of times. That means they can not predict a change or alter clinical outcomes. The reality doesn't listen to these conjectures.
Dr Lynn and I postulate that the original, upstream mistake lies in the very heuristic used to lump together many different diseases under umbrella terms such as ARDS and sepsis. The lumpers believe that similar presentations indicate similar mechanisms. The consequence is that each RCT is a unique mix of different biological mechanisms testing one specific mechanistic target. Even in the case of existing effects on one or a few patients, it gets diluted by a large, in fact unknown, number of patients whose clinical presentation doesn't share the same pathophysiology.

Whether one can deduce the cause from the effects has been a major issue in epistemology for generations. The pendulum is now swinging towards the affirmative, at least in Medicine, and mainstream medical researchers confidently preach to uninformed audiences that advanced analytical methods can ascertain causality by observing visible data. I don't share their beliefs. Their analysis is always context-dependent, hence provisional.
Anyway, this is for another post. My point today is that researchers are trying to refine their empiricism by sorting septic patients by phenotypes. Ongoing projects select subjects using a lumping strategy, i.e., Sepsis-3 criteria, and then divide the patients phenotype-wise to test a phenotype-related intervention. By doing that, they think they are splitting what was erroneously lumped by the lumping criteria/definition. Not so fast, escalophobetic researcher!
You can't attribute causality to a phenotype because you don't have the counterfactual, in other words, the expression of the phenotype in non-septic individuals.

Again. If we plan to stick to the dysregulated inflammation paradigm, we shouldn’t study only patients with dysregulated inflammation. We must measure the prevalence of the phenotype in the non-exposed.
Let's keep the discussion within the paradigm that describes sepsis as organ dysfunction caused by dysregulated inflammation in response to infection. A response phenotype may be a change in the expression of a soluble biomarker, a receptor, or the activity of an enzyme, etc. A non-exposed subject may be one with pairing baseline characteristics but without infection, or with infection but lacking sepsis criteria.
Triaged-out patients bear essential information
Case study: purinergic receptor expression in sepsis
Purinergic signaling is integral to pro- and anti-inflammatory responses. Extracellular adenosine triphosphate (ATP) is the best studied pro-inflammatory purine. It acts as a DAMP (damage-associated molecular pattern) to promote inflammation, mainly through P2 receptors present in polymorphonuclear leukocytes. Excessive ATP signaling is curbed by membrane-bound ATPases, CD39 and CD73, but also by soluble phosphatases. The phosphatases convert ATP into adenosine, a molecule with anti-inflammatory properties that is mediated by adenosine receptors. Serum ATP and adenosine levels, coupled with the relative expression of ATP (P2) / adenosine (A) receptors, offer a molecular “switch” between hyperinflammation (SIRS) and immunosuppression (CARS).
It stems from the dysregulated inflammation paradigm that septic subjects are expected to have some differential expression of P2 receptors, and they do (figure). Moreover, if you look only at the “septic” bars, you see that some septic subjects express more P2 receptors than others. Septic subjects differ. Got it! All you need to do is place a dividing threshold related to your favorite clinical outcome, and there is your sepsis “High P2 receptor expression” phenotype.
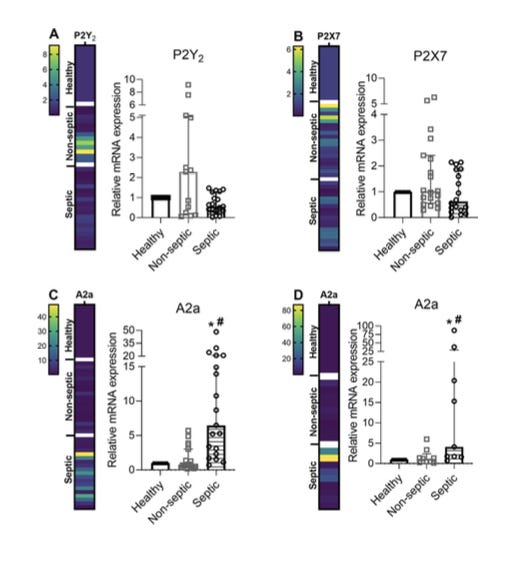
However, does it relate to being septic? You can only figure it out if you also examine non-exposed (non-septic) individuals. The “non-septic” bar is frustrating. There appears to be a large overlap, and intriguingly, P2 receptors seem to be less expressed in septic patients.
How valid is your sepsis phenotype if it is not even firmly related to sepsis?
I think it makes the case. All current “sepsis phenotypes” RCTs lack internal validity, because researchers don't even know whether the phenotypes are related to sepsis. It is a downstream consequence of the Petty and Bone mistake.
The only way forward is to drop the sepsis definitions in favor of a phenotype-wise approach. This is the necessary approach if we remain within the dysregulated inflammation paradigm. It will be difficult, but the alternative is persist in failure mode.
Thank you for reading The Thoughtful Intensivist.
Dr Lynn has recently made a strong case of the Petty and Bone mistake. Please read it here.
Interested in purinergic signaling? Please read the article here. If you can’t open it, I’ll be happy to send you a PDF.
Click below to share or subscribe to the most insightful and self-assured voice of critical care.
The counterfactual argument about phenotypes is sharp. Ive seen this same oversight in machine learning research where people build models on selected populations then try to generalize findings withot validating against unselected cohorts. The point about triaged-out patients containing essential information really cuts to the core of selection bias. Seems like every field has its version of the Petty and Bone mistake, where operational convenience slowly transforms into epistemic certainty.
I had to read it twice. But I think i got it. Whats the alternative definition? If we disregard the nonsense term dysregulated host response how do we define host sepsis moving forward?